科(kē)技(jì )有(yǒu)限公(gōng)司")
科(kē)技(jì )有(yǒu)限公(gōng)司")
術服務(wù)")

16sRNA腸道微生物(wù)多(duō)樣性
所屬分(fēn)類:
實驗介紹
原核微生物(wù)的16S rRNA、真菌的ITS基因、18S rRNA基因或特定的功能(néng)基因能(néng)夠用(yòng)于微生物(wù)的分(fēn)類鑒定和系統發育研究,廣泛應用(yòng)于微生物(wù)生态學(xué)研究中(zhōng)。近年來随着高通量測序技(jì )術及數據分(fēn)析方法的不斷進步,大量基于這些基因的研究使得微生物(wù)生态學(xué)得到了迅速發展。本項目采用(yòng)Illumina測序技(jì )術的增子測序,具(jù)有(yǒu)高通量和高精(jīng)度等優點,目前已成為(wèi)研究環境微生物(wù)群落組成和多(duō)樣性的主流方法。
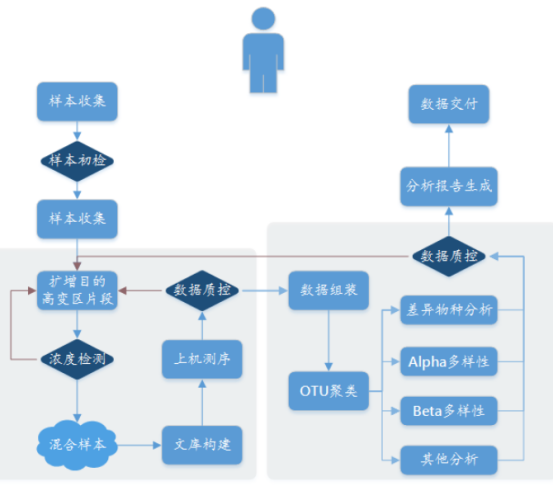
實驗流程
提供結果
測序得到的原始下機數據經過拼接,過濾得到可(kě)供後續分(fēn)析的高質(zhì)量目标序列。後續生物(wù)信息學(xué)操作(zuò)使用(yòng)Usearch(http://www.drive5.com/usearch)和QIIME5等完成,統計和作(zuò)圖主要使用(yòng)R6、python和java等完成,主要操作(zuò)步驟概述如下:
1.序列拼接。
2.根據Barcode區(qū)分(fēn)樣品。
3.去除低質(zhì)量序列。
4.去除嵌合體(tǐ)。
5.在97%的相似性水平上進行OTU(Operational Taxonomic Units)的聚類。
6.挑選出OTU的代表性序列。
7.使用(yòng)物(wù)種分(fēn)類數據庫進行物(wù)種分(fēn)類信息的劃分(fēn),數據庫信息見主目錄下的README.docx。
8.對代表性序列進行比對并過濾,然後重構建進化樹。
9.過濾掉不需要的OTU并進行重抽樣。
10.計算各個分(fēn)類水平上的豐度信息。
11.群落組成分(fēn)析。
12.Alpha多(duō)樣性分(fēn)析。
13.Beta多(duō)樣性分(fēn)析。
14.差異物(wù)種分(fēn)析。
15.其它如物(wù)種間的相關性分(fēn)析等。
上一頁(yè): 無
下一頁(yè): 18S rDNA/ITS真菌多(duō)樣性
更多(duō)産(chǎn)品
鏡(TEM)")




生物(wù)")
如有(yǒu)問題可(kě)以留言給我們,我們将竭誠為(wèi)您服務(wù)。
*注:請務(wù)必信息填寫準确,并保持通訊暢通,我們會盡快與你取得聯系
四川賽因斯特生物(wù)科(kē)技(jì )有(yǒu)限公(gōng)司
全國(guó)服務(wù)熱線(xiàn):028-67172373
地 址:成都市溫江區(qū)金馬街(jiē)道海發路669号1棟5樓

官方微信公(gōng)衆号
浏覽手機網站
CopyRight © 2024 四川賽因斯特生物(wù)科(kē)技(jì )有(yǒu)限公(gōng)司 版權所有(yǒu)
在線(xiàn)咨詢
SAF Coolest v1.3.1.1 設置面闆 HNFSD-ZFSY-IWXAE-SQW
無數據提示
Sorry,當前欄目正在更新(xīn)中(zhōng),敬請期待!
您可(kě)以查看其他(tā)欄目或返回 首頁(yè)